Alix Lasers ® certificates
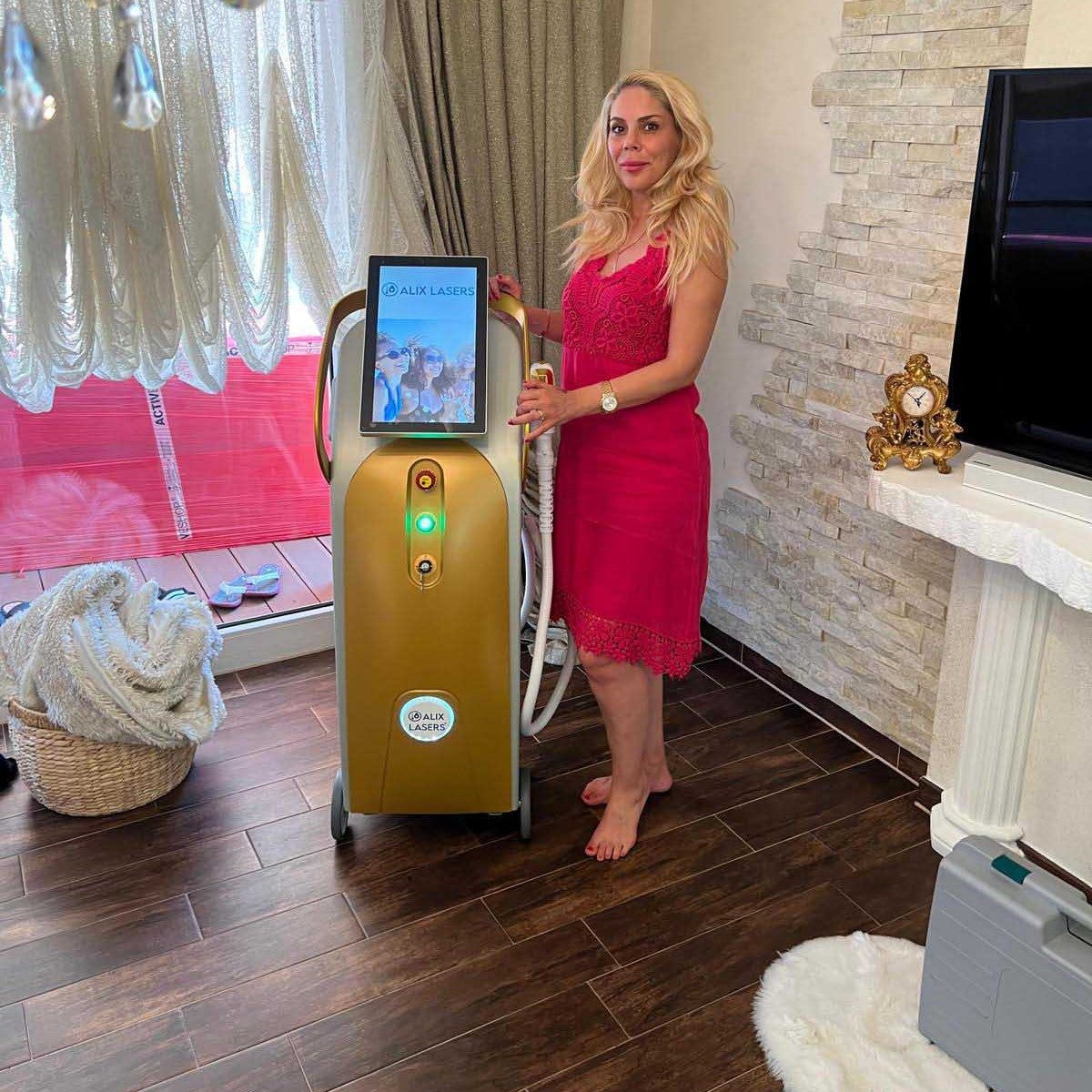
Alix Lasers ® devices are subject to the highest safety standards and have worldwide approvals for safe operation.
TÜV Süd all diode lasers
The path to the different markets is paved with strict requirements, especially in the area of medical devices: The products must meet the highest quality standards and their quality management must be designed accordingly. Here it makes sense and is good to be able to consult an experienced expert. Just like TÜV SÜD.
TÜV SÜD has been checking and certifying quality management for medical devices for over 30 years and has the necessary know-how, not least thanks to the constant further training of its specialists. That is why TÜV SÜD is one of the market leaders in countries with strict regulation of medical devices. Quality management certificates confirm compliance with a whole range of international standards - above all ISO 13485. As a manufacturer, you have proof that certified quality management that meets the highest requirements was used in the production of your medical devices. This promotes your image as a manufacturer, strengthens the trust of authorities, buyers and users in your products and provides clear sales arguments.
Medical devices manufactured or offered by companies that do not have a certified quality management system typically have a harder time gaining approval in major markets. This can lead to delays in market entry and loss of sales.
ISO 13485 test seal / test standard for all diode lasers
Certifications create trust – and this is particularly crucial for medical devices. The international standard EN ISO 13485 is the standard for quality management systems in the medical device sector. This certification is the prerequisite for market entry in many countries. Although it is an independent document, ISO 13485 is largely identical to ISO 9001. While ISO 9001 requires that the organization as a whole must strive for continuous improvement, the core claim of ISO 13485 relates to product safety. The aim here is to ensure that the product requirements are met through the effectiveness of the processes introduced. ISO 13485 contains detailed requirements on topics relating to the manufacture and marketing of medical devices.
Medical devices – test seal for all diode lasers
The EN ISO 13485 standard “Medical devices: Quality management systems – Requirements for regulatory purposes” addresses the requirements that medical device manufacturers and suppliers must meet when developing, implementing and maintaining management systems for the medical device industry. The standard, originally developed in the 1990s, contains requirements for quality management systems that meet customer requirements, but also the regulatory requirements of the European Union (EU), Canada and other major markets worldwide.
EN ISO 13485 is similar to the ISO 9001 standard in its scope and purpose. However, it contains additional, specific requirements for medical devices and reformulates some of the requirements of ISO 9001. In most markets, certification according to ISO 9001 is therefore not an adequate replacement for certification according to the requirements of EN ISO 13485.
ECM – Duck Certification Macchine
Ente Certificazione Macchine is a well-known body authorized by the European Commission to issue EC certificates in accordance with Directive 93/42/EEC (MDD). [See the ECM accreditations on the European Commission's Nando Information System database]
The CE mark The CE mark is the mandatory certification mark that every medical device must have in order to be marketed and used in the European Union. The CE mark declares that the product complies with the applicable European directives. The Medical Devices Directive 93/42/EEC (MDD) lists the basic safety, effectiveness and quality requirements that a medical device must meet in order to receive the CE mark and be placed on the European market.
EU Regulation 765/2008
The CE marking indicates that the machine meets the requirements of the Machinery Directive. This also applies to all other applicable EC directives. Each machine may have exactly one CE mark. The CE marking must be placed on an equal footing with the manufacturer's information and must be affixed using the same technique.
CE stands for Communauté Européenne (European Community). The marking indicates that a product or machine complies with the relevant European guidelines. The CE mark, introduced in 1995, can therefore be understood as a “passport” for machines, as it allows machines to be both marketed and operated in the EU. It is important that the CE marking should not be confused with a seal of quality or a quality mark.
FDA
Food and Drug Administration (FDA) approval is a requirement to distribute medical devices in the United States; the other is to work with a qualified distribution partner.
Almost all medical device manufacturers supplying the American market must implement a quality management system that meets FDA QSR 21CFR820 or the Quality System Regulation. The requirements of the FDA QSR are similar to those of the European ISO 13485; At the same time, there are special features and the FDA does not recognize the ISO 13485 certificate.
Become part of the growing Alix Lasers® family